|
|
電子情報通信学会 - IEICE会誌 試し読みサイト
© Copyright IEICE. All rights reserved.
|
|
|
電子情報通信学会 - IEICE会誌 試し読みサイト
© Copyright IEICE. All rights reserved.
|

 | 学生/教養のページ Webアプリで体験するマテリアルズインフォマティクスの最先端 |
Molecular Generation Experience: A Web Application of Materials Informatics
気候変動や環境汚染,エネルギー資源の枯渇問題,食糧不足問題,そして未知の病原ウイルスなど,今日の我々の生活を取り巻く地球環境は,かつてないほど複雑で巨大な問題に直面している.レジ袋の有料化や二酸化炭素排出量の規制など世界中で様々な問題解決が試みられているが,その問題の大きさゆえに,必ずしも社会的な制度の変革だけで解消できるものではない.IBMリサーチ(基礎研究所)は,約半世紀にわたる材料科学とコンピュータ科学の研究開発の知見を生かし,材料開発の観点からもこのような問題解決へ向けて取り組んでいる.具体的には,二酸化炭素を吸着するポリマー,毒性の低い半導体加工用材料(フォトレジスト),エネルギー効率の高い太陽電池,リサイクル可能なペットボトルの開発など,産業の発達と地球環境の持続性を共にもたらす新材料の開発である(1).
これまで新材料の開発は,材料化学の専門家が各人の知識・経験・勘を頼りに,今日手に入る最も性能の良い(例:最も軽くて硬い,など)材料を少しずつ改良してゆくため,10年から20年もの歳月がかかるとされてきた.このように時間のかかる材料開発を大幅に加速するために,近年,データサイエンスや機械学習,AIを積極的に活用するマテリアルズインフォマティクス(MI)という分野が大きな注目を集めている.日本でも2015年にコンソーシアムMI2Iが発足されるなど,産官学連携で活動が進められている(2).MIで重要な技術領域は大きく分けて四つある.(1)文献からのデータ抽出,(2)物理化学シミュレーション,(3)化合物・構造デザイン,(4)自動化学実験,である.本稿では,中でも(3)化合物・構造デザイン領域における技術としてIBMリサーチが開発したオープンプラットホーム「IBM Molecule Generation Experience(MolGX)」について紹介する.
MIにおいて特に注目を集める技術である,分子構造をAIによりデザインする分子生成モデルと,MI全般における一般ユーザがアクセス可能なプラットホームの現状について紹介する.
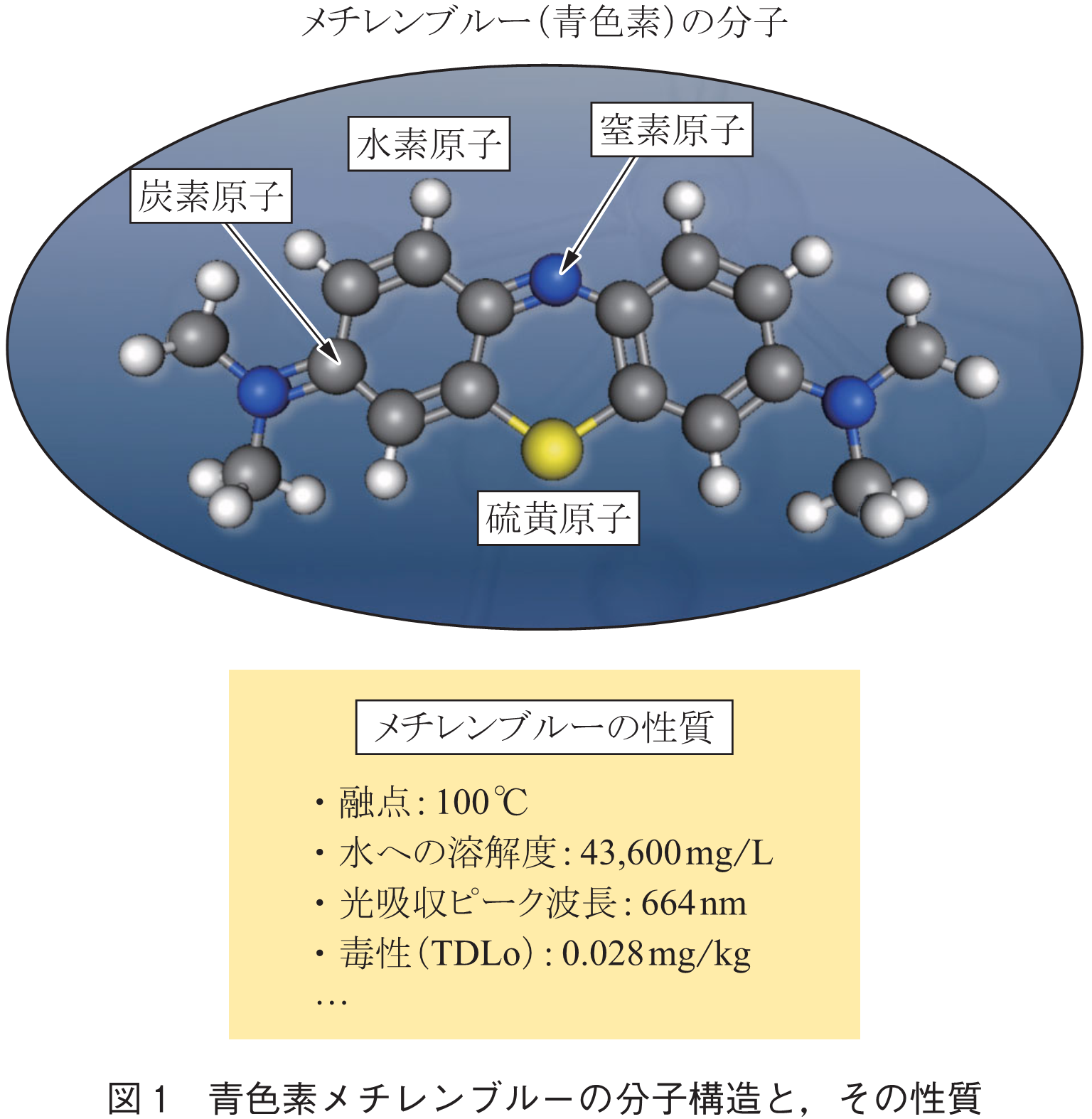
あらゆる材料の性質は,材料を構成する原子の組合せ方によって決定される.ポリマーや色素のような有機材料は,分子の集合によって成るため,その分子の構造つまり原子の組合せを適切に設計(デザイン)することで,硬さや色,水への溶けやすさなど欲しい性質を実現することができる.図1に,青色素メチレンブルーの例を挙げている.近年,生成モデルと呼ばれるAI技術を分子デザインに活用する試みがなされている.生成モデルとは,AIを使って絵や音楽,文章を自動的に生成する技術の総称で,変分オートエンコーダ(VAE: Variational Auto Encoder)や敵対的生成モデル(GAN: Generative Adversarial Network)などがそれに当たる.近年の分子生成モデルのトレンドは,ディープニューラルネットワーク(DNN: Deep Neural Network)で構成されたVAEやGANによるテキスト生成技術を分子構造の生成に応用するものである.あらゆる分子は,炭素や窒素などの原子同士が単結合や二重結合を介してつながったグラフ構造を持つが,そのグラフ構造は,Simplified Molecular Input Line Entry System(SMILES)と呼ばれる文法にのっとった文字列により表記できる.例えばカフェインの分子構造は,SMILES文法にのっとるとCN1C=NC2=C1C(=O)N(C(=O)N2C)Cと表記することができる.アルファベットは元素記号,数字は環構造を閉じる位置,括弧は枝分かれを示している.テキスト生成用に構成されたVAEやGANをベイズ最適化や強化学習などと組み合わせることで,欲しい性質を持つ分子構造を表現するSMILES候補を自動的に列挙することができる(3).またSMILES以外でも,分子グラフを隣接行列によって表現する手法(4)やノードの連結されたグラフ構造として直接表現する手法(5)などもあり,いずれも構造生成にDNNを用いている.
DNNを用いる生成モデルはAI技術として高い新規性が認められるものの,実際のユーザである実験化学者の立場からは実用上の課題がある.まず,DNNの性質上,モデルの解釈(「なぜそのような結果が出てきたか」)を得ることができない.また,分子構造の情報を適切に変換し読み込む機能(エンコーダ)と構造生成の機能(デコーダ)がDNNによって構成されているため,膨大なデータや複雑なパラメータチューニング,長時間にわたるDNNモデルの学習が必要であり,材料化学の現場にとって利用のハードルが非常に高い.このような現状に鑑み,筆者らはDNNを用いず,エンコーダとデコーダ部分の機能をグラフ理論に基づくアルゴリズムにより事前に作り込んだ,新しいタイプの分子生成モデルを開発した.
上記のような実用性を考慮したツール開発に加え,マテリアルズインフォマティクスやAI分野全般のオープンイノベーションや,幅広い一般普及・教育的な観点からも,これらの技術を一部の研究者・開発者コミュニティ内だけで閉じるのではなく,オープンプラットホーム上で公開し多くの人々に使ってもらうことが重要である.しかしながら,今日一般公開されているほとんどのMI技術はGitHub上におけるソースコードの開示であるため,一定レベルのコーディングやITスキルが必要とされ,データサイエンティストやAI研究者は利用可能であっても,これらのツールの将来における実際のユーザであるはずの実験化学者が容易に利用できるとは言い難い.一方,誰にでも利用しやすいグラフィカルユーザインタフェース(GUI)を備えたMIのWebアプリケーションも,少数ながら存在する.
例えばStarrydata2(6)は,ユーザが文献データをアップロードすることで,文献内のグラフ情報を自動的に読み取り,オープンデータベース化してくれる.文献からのテキストデータ抽出・ナレッジグラフによる仮説生成ツールとして,Corpus Conversion ServiceやCorpus Processing Serviceがある(7).Polymer Genomeでは,入力したモノマー構造から予測されるポリマーの物性値を簡単に得ることができる(8).また,RXN for Chemistryでは,入力した化合物の化学反応や合成経路を予測することができる(9).
本稿で紹介するMolGXは,これらWebアプリと同様のシンプルなGUIにより,分子生成モデルの機能を一般ユーザに提供する世界初のサービスである.
ここでは,MolGXの背景技術について,実用向けの「プロフェッショナル版」をベースに紹介する.MolGXのアルゴリズムの基本的な考え方は,順問題と逆問題のペアである.基本的なワークフローは,(0)データ入力,(1)特徴エンコード,(2)予測モデル作成,(3)特徴ベクトル候補列挙,(4)構造生成,から成る(図2).詳しい説明は文献(10)を参照頂きたい.以下で,各ステップについて説明する.
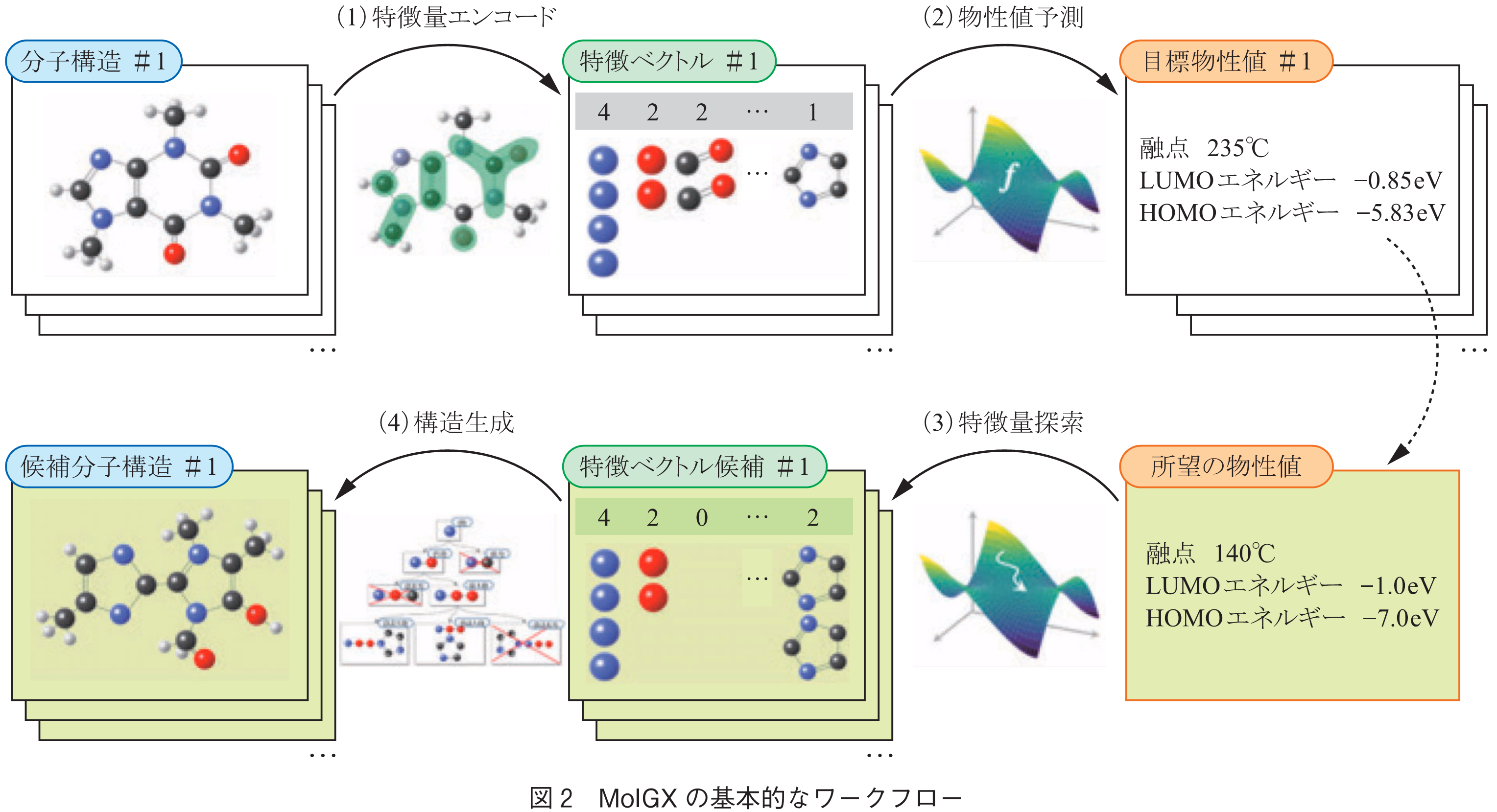
学習データは,SMILESによって表記された分子構造と,その分子が持つ物性値のペアから成るCSVフォーマットで与えられる.物性値は複数設定することができる.分子構造のみならず,測定条件や重合条件(モノマーの場合)なども含めることができる.なおここでSMILESを用いているのは,CSVファイル内で分子を表現するための便宜的な理由によるものであり,先に述べたSMILES生成モデルとは関係ない.読み込んだSMILESはMolGX内でノード・エッジ情報を持つ分子グラフデータに変換される.
分子グラフ構造は,特徴量(特徴ベクトル)に変換される.MolGXでは,グラフカーネルと呼ばれる考え方に基づき,主に部分構造すなわち分子グラフを構成するパーツの個数として,各特徴量を表現する.例えば,ある分子が炭素原子を3個,窒素原子を2個,部分構造C=Cを1個,部分構造C-Nを1個,・・・を含む場合,特徴ベクトルは[3,2,1,1,…]として得られる.どのような部分構造をカウントするかは,ユーザが部分構造のサイズを指定すれば,あとはMolGXが入力された分子構造データを基に,カウントすべき部分構造を自動的に定義する.分子構造に併せて測定条件なども入力情報として与えた場合は,これらも特徴ベクトルの一部として連結される.
得られた特徴ベクトルを用いて,物性値を予測するための回帰モデルを作成する.ここでは通常の機械学習による回帰モデル(ラッソ回帰・リッジ回帰など)を使用する.ここまでが,分子構造を与えるとその物性値を予測する過程,すなわち「順問題」と呼ばれる過程である.
ここからは,逆問題を解くプロセスである.まず,3.3で得られた予測モデルを基に,ユーザが欲しい物性値を持つような特徴ベクトルの候補を探索し,列挙する.ここではヒューリスティックな最適化アルゴリズム(粒子群最適化)を用いて解探索する.ここで重要なことは,化学構造として意味のある(化学構造に変換し得る)特徴ベクトルは,数十次元にわたる解空間上の非常に限られた多様体上にしか存在しないということである.たとえ予測モデル上で所望の物性値を持つ特徴ベクトルが得られたとしても,ほとんどの場合,各ベクトル要素(つまり部分構造の個数)同士に矛盾が生じ,化学構造にデコードすることができない.したがって,MolGXでは部分構造の包含関係などを考慮して解空間に自動的に制約を与える仕組みを持つ.
特徴ベクトルの候補が得られたら,各特徴ベクトルを具体的な化学構造に「デコード」する.特徴ベクトルの各要素は部分構造の個数によって表現されるため,これらを効率良く連結させるグラフ列挙アルゴリズムにより構造生成する.構造を漏れなくかつ重複なく生成するために,Mckay’s Canonical Construction Pathというグラフ理論を基礎とする数理アルゴリズムをベースに化学構造用に改良を重ね,化学的知見に準じた様々なスクリーニングを設けることで,高速な構造生成を実現した.
これまで見てきたとおり,DNNとは違い,特徴量エンコード及び構造生成部分のワークフローがアルゴリズムにより事前に明確に定義されているため,この箇所を大量のデータを用いたDNN学習によりユーザが作り込む必要がない.また,特徴量の内容が明らかかつ,部分構造を連結させてゆく構造生成のプロセスを逐一追うことができるので,得られたモデルの精度や生成された分子構造に対する理由付けや解釈を得ることができるといった利点を持つ.
筆者らは前章で紹介した「プロフェッショナル版」アルゴリズムをPythonパッケージとして実装した.コアとなるアルゴリズム以外にも,化学構造の前処理・後処理,ワークフローの制御など実用上の機能など約200種類のメソッドを含む.本ツールは実験化学者らによる現場での利用を目的に作成したものであり,様々な企業で利用されている(11).筆者らは「体験版」としてその一部の機能を切り出して直観的なユーザインタフェースを実装することで,一般ユーザでも無料で利用できるWebアプリケーションとして公開した.AIや化学の初学者でも簡単に利用できるよう,ワークフローを①データの観察・選択,②予測モデル学習,③構造生成,の3ステップに簡略化し,これに合わせてツールの各メソッド約20種類をAPI化した.以下に,それぞれのステップを紹介する.
備え付けのデータセット(QM9,ZINC15)から利用したいデータセットを選択する.データは分子構造と,それにひも付くエネルギーギャップ(分子が吸収できる光の波長に相当),熱容量(温まりやすさの指標),LogP(脂溶性の指標),など基本的な物性値により構成される.図3(a)に示すような散布図上で物性値の分布及び分子構造を確認し,その中からモデルの学習に使用するデータのみをサブセットとして選択する.
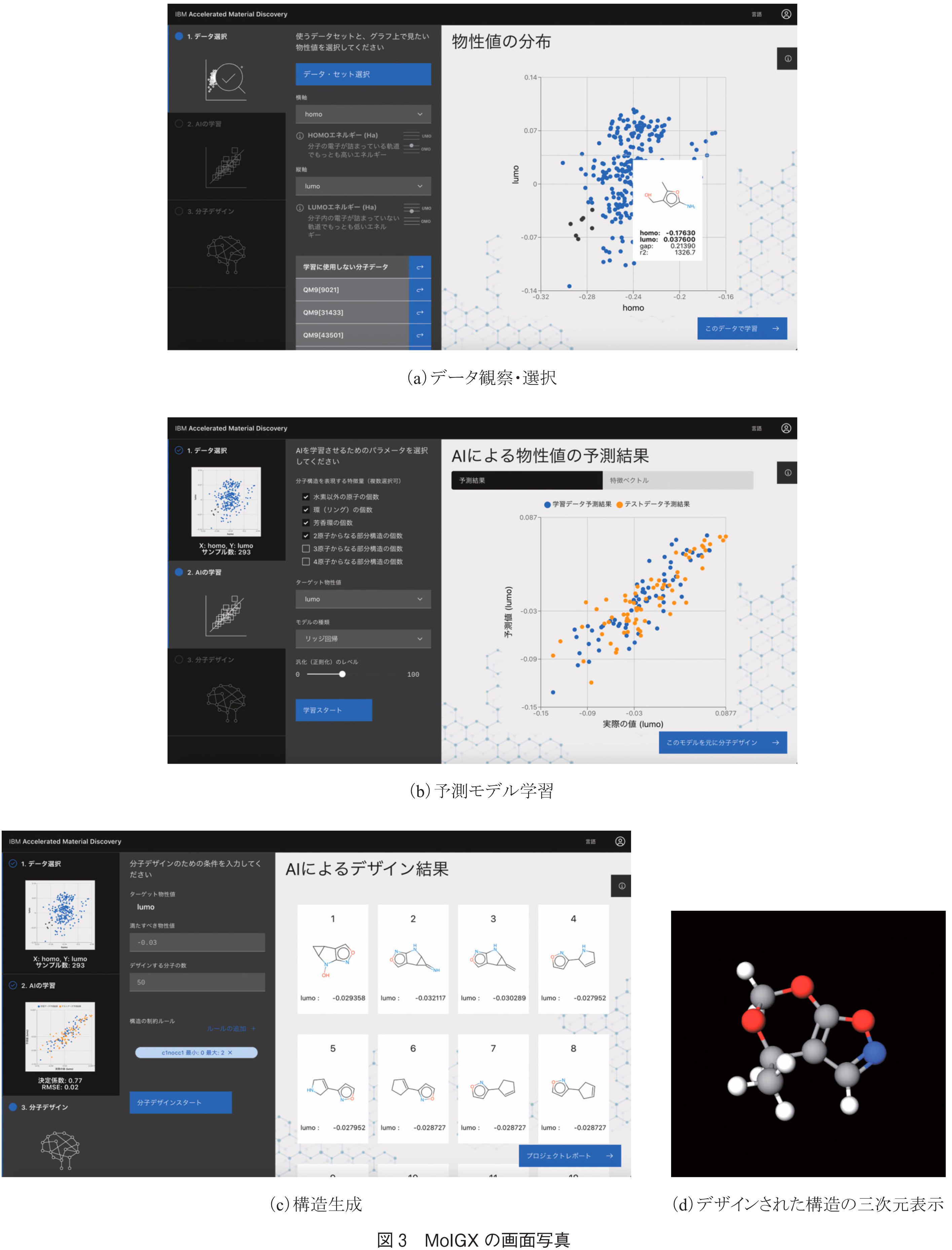
前ステップで選択したサブセットを用いて,物性値を予測するモデルを学習させる.学習に際し,使用する特徴量の種類(カウントすべき部分構造のタイプなど),予測したい物性値,モデルの種類(リッジ回帰,カーネルリッジ回帰など),汎化レベル,を設定する.汎化つまり正則化のパラメータは本来複数種類あるが,本WebアプリはAI・機械学習の考え方を理解する教育目的も兼ねるため,簡単のため一つのパラメータにまとめている.
図3(b)に示したとおり,学習結果はグラフとして表示されるほか,特徴ベクトルの中身を確認することもできる.
前ステップで学習した予測モデルを基に,目標とする物性値,生成する個数,構造の制約条件(-OHは3個以下にする,など)を指定し,構造生成をスタートする.目標物性値が元のデータ範囲内にある方が生成しやすい.QM9を使用した場合,100個生成するのに20秒から1分以内に完了することが多い.生成された分子構造は一覧表示で確認できるほか,レポートページでは構造を三次元表示し,回転させるなど「触れる」ことができる(図3(c)(d)).
上記ツールは,スケーラブルなアーキテクチャを備えたWebアプリケーションとして,KubernetesによりIBMのハイブリッドクラウド上に実装された(https://molgx.draco.res.ibm.com/).ユーザは,Twitter,Facebook,Googleアカウントのいずれかを用いてログインすることができる.また,全ページにつき日本語と英語の両方をサポートしており,誰もが世界中から簡単に分子生成モデルを利用することができる.
IBMリサーチの開発した分子生成モデル「IBM Molecule Generation Experience(MolGX)」の概要及び,そのWebアプリケーションについて紹介した.このようにツールの一部の機能をWebアプリとして公開したのは,AIやマテリアルズインフォマティクスといった新技術の潮流が押し寄せる中,学生の皆さんをはじめとする本分野の初学者や興味のある人々にいち早くその技術に触れ,考え方を理解してもらうことで,次世代の科学技術の発展へ向けて社会全体で備えたいという動機によるものである.MolGXは今後もサイエンスプラットホームとして進化してゆく.ここに,材料分野のオープンプラットホームといったサイエンスの新たな可能性を感じて頂けたら,チーム一同幸いである.
(1) THINK Blog Japan, “IBM 5in5:新たな材料の発見プロセスの大幅な加速によって実現する持続可能な未来.”2020.
https://www.ibm.com/blogs/think/jp-ja/ibm-5-in-5-accelerating-process-of-discovery/
(2) 武田征士,濱 利行,徐 祥瀚,山根敏志,中野大樹,“新材料探索におけるAIとデータの活用,”信学誌,vol.103, no.6, pp.621-628, June 2020.
(3) R. Gómez-Bombarelli, J.N. Wei, D. Duvenaud, J.M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T.D. Hirzel, R.P. Adams, and A. Aspuru-Guzik, “Automatic chemical design using a data-driven continuous representation of molecules,” ACS Central Science, vol.4, no.2, pp.268-276, 2018.
(4) N. De Cao and T. Kipf, “MolGAN: An implicit generative model for small molecular graphs,” arXiv: 1805.11973[stat. ML], 2018.
(5) W. Jin, R. Barzilay, and T. Jaakkola, “Junction tree variational autoencoder for molecular graph generation,” Proc. 35th International Conference on Machine Learning, PMLR 80: 2323-2332, 2018.
(6) Starrydata2.
https://www.starrydata2.org/
(7) Corpus Conversion Service.
https://research.ibm.com/covid19/deep-search/
(8) Polymer Genome.
https://www.polymergenome.org/
(9) IBM RXN for Chemistry.
https://rxn.res.ibm.com/
(10) S. Takeda, T. Hama, H.-H. Hsu, V.A. Piunova, D. Zubarev, D.P. Sanders, J.W. Pitera, M. Kogoh, T. Hongo, Y. Cheng, W. Bocanett, H. Nakashika, A. Fujita, Y. Tsuchiya, K. Hino, K. Yano, S. Hirose, H. Toda, Y. Orii, and D. Nakano, “Molecular inverse-design platform for material industries,” Proc. 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, pp.2961-2969, Aug. 2020.
https://www.kdd.org/kdd2020/accepted-papers/view/molecular-inverse-design-platform-for-material-industries
(11) 長瀬産業株式会社,“TABRASA.”
https://tabrasa.jp/
(2021年3月31日受付)

オープンアクセス以外の記事を読みたい方は、以下のリンクより電子情報通信学会の学会誌の購読もしくは学会に入会登録することで読めるようになります。 また、会員になると豊富な豪華特典が付いてきます。
電子情報通信学会 - IEICE会誌はモバイルでお読みいただけます。
電子情報通信学会 - IEICE会誌アプリをダウンロード

![]()